Abstract
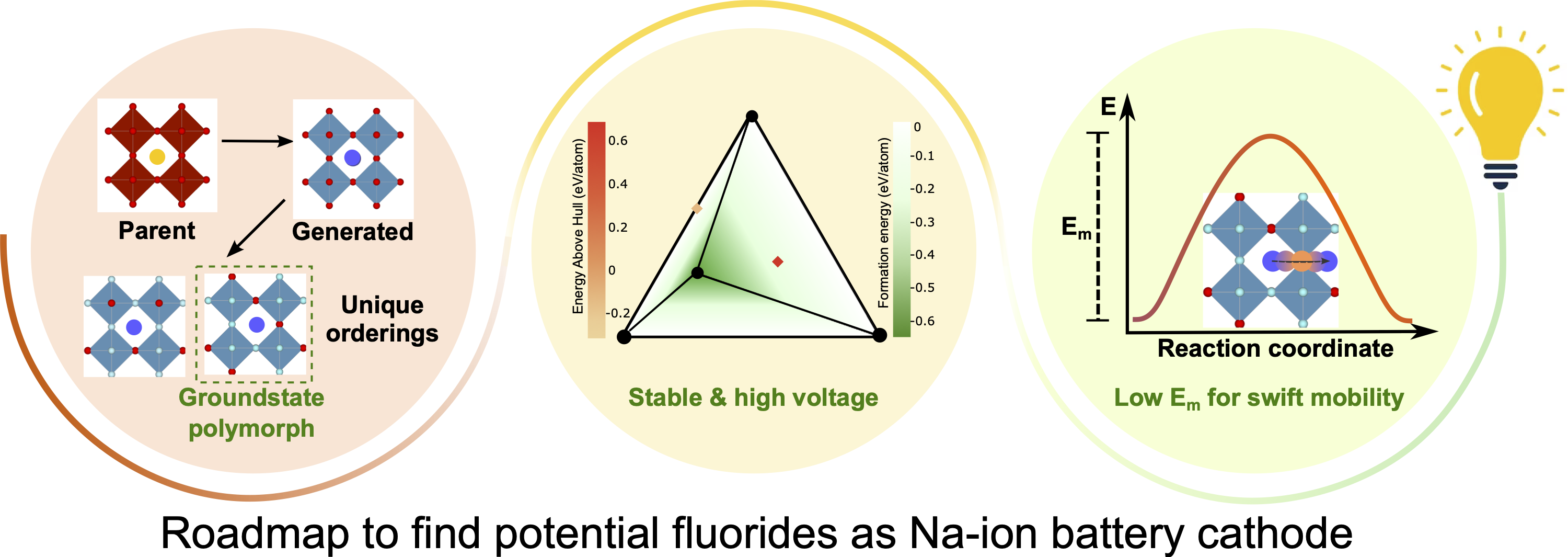
Na-ion batteries (NIBs) are increasingly considered as a viable alternative to Li-ion batteries due to the abundance, low cost, and thermal stability of Na-based systems. To improve the practical utilization of NIBs in applications, it is important to boost the energy and power densities of the electrodes being used by the discovery of novel candidate materials. Thus, we explore the chemical space of transition metal-containing oxyfluorides (TMOFs) that adopt a perovskite structure as possible NIB electrodes. Our choice of the perovskite structure is motivated by the “large” cationic tunnels that can accommodate Na+, while the chemistry of TMOFs is motivated by the high electronegativity and inductive effect of F−, which can possibly lead to higher voltages. We use density functional theory-based calculations to estimate the ground state polymorphs, average Na (de)intercalation voltages, thermodynamic stabilities, and Na+ mobility on two distinct sets of compositions: the F-rich NaxMOF2 and the O-rich Na1+xMO2F, where x = 0−1 and M = Ti, V, Cr, Mn, Fe, Co, or Ni. Upon identifying the ground state polymorphs in the charged compositions (i.e., MOF2 and NaMO2F), we show that F-rich perovskites exhibit higher average voltages compared to those of the O-rich perovskites. Also, we find six stable/metastable perovskites in the F-rich space, while all the O-rich perovskites (except NaTiO2F) are unstable. Finally, our Na-ion mobility calculations indicate that TiOF2−NaTiOF2, VOF2−NaVOF2, CrOF2, and NaMnOF2 can be promising compositions, albeit with challenges to be resolved, for experimental exploration as NIB cathodes. These oxyfluoride compositions can be promising if used primarily in a strained electrode configuration and/or in thin film batteries. Our computational approach and findings provide insights into developing practical NIBs involving fluorine-containing intercalation frameworks.