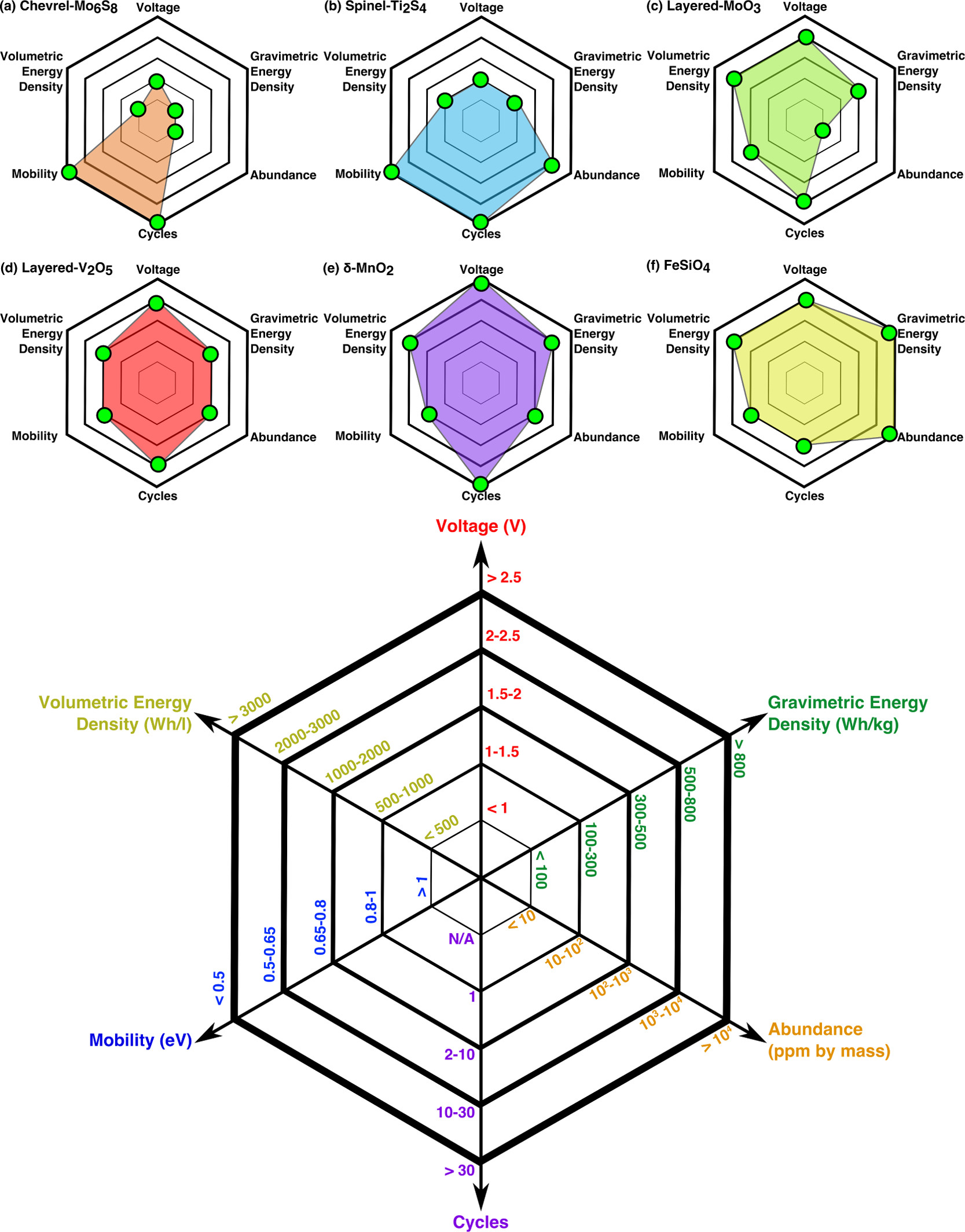
Batteries, especially with the advent of the state-of-the-art Li-ion architecture, have become ubiqutous as energy storage devices for a range of applications, from portable electronics and electric vehicles to grid-scale storage. However, given the supply-chain and geo-political constraints associated with some of the elements critical in making the modern Li-ion battery, it is imperative to explore frameworks that can achieve similar energy densities as Li-ion with more earth-abundant elements. Promising beyond Li-ion architectures include the development of Na-ion and multivalent (based on Mg-ion for example) batteries.
All battery frameworks that go beyond Li-ion require robust materials, either as electrodes that can “intercalate” the active ion reversibly, and/or electrolytes that are stable against both electrodes. Specifically, solid electrolytes are of significant interest in the development of beyond Li-ion batteries owing to their safety. Another important requirement in developing new battery frameworks is to develop better interfaces (or coating layers) that exist between electrodes and electrolytes. Thus, this field of research will include discovery of new materials, such as high-voltage negative electrodes, robust ionic conductors and stable interfaces, while also understanding the behavior of existing candidates, using density functional theory (and beyond) calculations.
References
- Canepa et al., “Odyssey of multivalent cathode materials: open questions and future challenges”, Chem. Rev. 117, 4287-4341 (2017). DoI.
- Sai Gautam et al., “Influence of inversion on Mg mobility and electrochemistry in spinels”, Chem. Mater. 29, 7918-7930 (2017). DoI.
- Butler et al., “Designing interfaces in energy materials applications with first-principles calculations”, npj Comput. Mater. 5, 19 (2019). DoI.