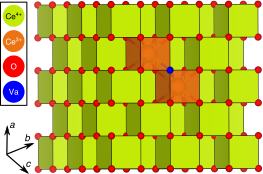
Abstract
Theoretical models that reliably can predict off‐stoichiometry in materials via accurate descriptions of underlying thermodynamics are crucial for energy applications. For example, transition‐metal and rare‐earth oxides that can tolerate a large number of oxygen vacancies, such as CeO2 and doped CeO2, can split water and carbon dioxide via a two‐step, oxide‐based solar thermochemical (STC) cycle. The search for new STC materials with a performance superior to that of state‐of‐the‐art CeO2 can benefit from predictions accurately describing the thermodynamics of oxygen vacancies. The sub‐lattice formalism, a common tool used to fit experimental data and build temperature‐composition phase diagrams, can be useful in this context. Here, sub‐lattice models are derived solely from zero‐temperature quantum mechanics calculations to estimate fairly accurate temperature‐ and oxygen‐partial‐pressure‐dependent off‐stoichiometries in CeO2 and Zr‐doped CeO2. Physical motivations for deriving some of the “excess” sub‐lattice model parameters directly from quantum mechanical calculations, instead of fitting to minimize deviations from experimental and/or theoretical data, are identified. Important limitations and approximations of the approach used are specified and extensions to multi‐cation oxides are also suggested to help identify novel candidates for water and carbon dioxide splitting and related applications.