Abstract
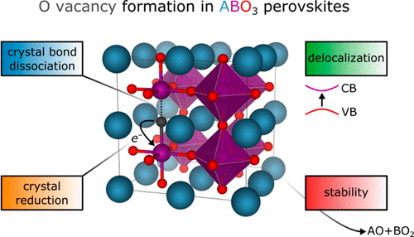
The control of oxygen vacancy (VO) formation is critical to advancing multiple metal-oxide-perovskite-based technologies. We report the construction of a compact linear model for the neutral VO formation energy in ABO3 perovskites that reproduces, with reasonable fidelity, Hubbard-U-corrected density functional theory calculations based on the state-of-the-art, strongly constrained and appropriately normed exchange-correlation functional. We obtain a mean absolute error of 0.45 eV for perovskites stable at 298 K, an accuracy that holds across a large, electronically diverse set of ABO3 perovskites. Our model considers perovskites containing alkaline-earth metals (Ca, Sr, and Ba) and lanthanides (La and Ce) on the A-site and 3d transition metals (Ti, V, Cr, Mn, Fe, Co, and Ni) on the B-site in six different crystal systems (cubic, tetragonal, orthorhombic, hexagonal, rhombohedral, and monoclinic) common to perovskites. Physically intuitive metrics easily extracted from existing experimental thermochemical data or via inexpensive quantum mechanical calculations, including crystal bond dissociation energies and (solid phase) reduction potentials, are key components of the model. Beyond validation of the model against known experimental trends in materials used in solid oxide fuel cells, the model yields new candidate perovskites not contained in our training data set, such as (Bi,Y)(Fe,Co)O3, which we predict may have favorable thermochemical water-splitting properties. The confluence of sufficient accuracy, efficiency, and interpretability afforded by our model not only facilitates high-throughput computational screening for any application that requires the precise control of VO concentrations but also provides a clear picture of the dominant physics governing VO formation in metal-oxide perovskites.