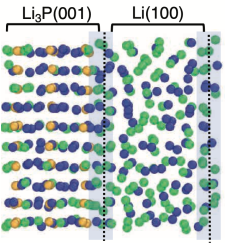
Abstract
A crucial ingredient in lithium (Li) and sodium (Na)-ion batteries (LIBs and NIBs) is the electrolytes. The use of Li-metal (Na-metal) as anode in liquid electrolyte LIBs (NIBs) is constrained by several issues including thermal runway and flammability, electrolyte leakage, and limited chemical stability. Considerable effort has been devoted toward the development of solid electrolytes (SEs) and all-solid-state batteries, which are presumed to mitigate some of the issues of Li-metal(Na-metal) in contact with flammable liquid electrolytes. However, most SEs, such as Li3PS4, Li6PS5Cl and Na3PS4 readily decompose against the highly reducing Li-metal and Na-metal anodes. Using first-principles calculations we elucidate the stability of more than 20 solid‖solid interfaces formed between the decomposition products of Li3PS4, Li6PS5Cl (and Na3PS4) against the Li-metal (Na-metal) electrode. We suggest that the work of adhesion needed to form a hetereogenous interfaces is an important descriptor to quantify the stability of interfaces. Subsequently, we clarify the atomistic origins of the resistance to Li-ion transport at interfaces of the Li-metal anode and selected decomposition products (Li3P, Li2S and LiCl) of SEs, via a high-fidelity machine learned potential (MLP). Utilising an MLP enables nano-second-long molecular dynamics simulations on `large’ interface models (here with 8320 atoms), but with similar accuracy to first-principles approaches. Our simulations demonstrate that the interfaces formed between Li-metal and argyrodite (e.g., Li6PS5Cl) decomposition products are resistive to Li-ion transport. The implications of this study are important since binary compounds are commonly found in the vicinity of Li (Na)-metal upon chemical and/or electrochemical decomposition of ternary and quaternary SEs.