Abstract
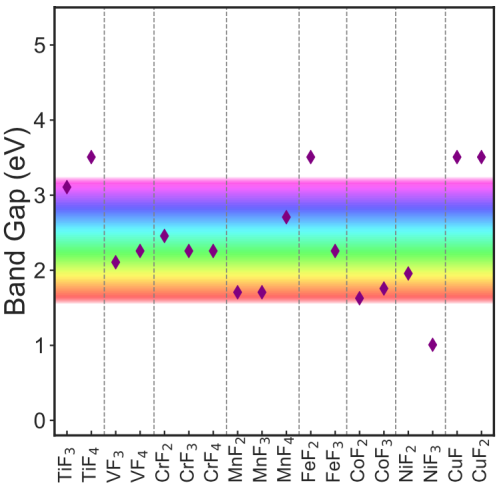
Accurate predictions of material properties within the chemical space of transition metal fluorides (TMFs), using computational frameworks such as density functional theory (DFT), is important for advancing several technological applications. The state-of-the-art semilocal exchange-correlation functionals within DFT include the strongly constrained and appropriately normed (SCAN) and the restored regularized SCAN (r2SCAN), both of which are meta generalized gradient approximation (metaGGA) functionals. Given their semilocal nature, both SCAN and r2SCAN are susceptible to self-interaction errors (SIEs) while modeling highly correlated d electrons of transition metals. Hence, in this work, we evaluate the accuracy of both SCAN and r2SCAN functionals in estimating several properties of TMFs, including redox enthalpies, lattice geometries, on-site magnetic moments, and band gaps. Specifically, we consider binary fluorides of Ti, V, Cr, Mn, Fe, Co, Ni, and Cu. We observe both SCAN and r2SCAN exhibit poor accuracy in estimating fluorination enthalpies among TMFs, which can be primarily attributed to SIEs among the d electrons, given both functionals bind F2 accurately. Thus, we derive optimal Hubbard U corrections for both functionals based on experimental fluorination (or oxidation) enthalpies within binary TMFs. Note that our attempts at using the linear response theory to derive U corrections yielded unphysical values for V, Fe, and Ni fluorides. While adding the fluorination-enthalpy-derived U corrections to the metaGGA functionals does not significantly affect the lattice volumes and on-site magnetic moments (and in turn, the accuracy of these property estimations versus experiments), it does cause a significant increase in calculated band gaps. Note that the U-corrected band gaps in several fluorides deviate to a lesser extent from band gaps calculated with a hybrid functional compared to the non-U-corrected functionals. Also, we calculated the average Na intercalation voltage in Mn, Fe, Co, and Ni fluorides, and stabilities of Na-V-F, Na-Cr-F, Na-Mn-F, and Na-Fe-F ternary compounds as transferability checks of our optimal U values. Overall, we do recommend the incorporation of the Hubbard U correction to improve predictions of redox enthalpies in other TMFs. Finally, our study should advance the accuracy of DFT-based screening studies to unearth novel TMFs, which can be used in various applications, including energy storage, catalysis, and magnetic devices.