Abstract
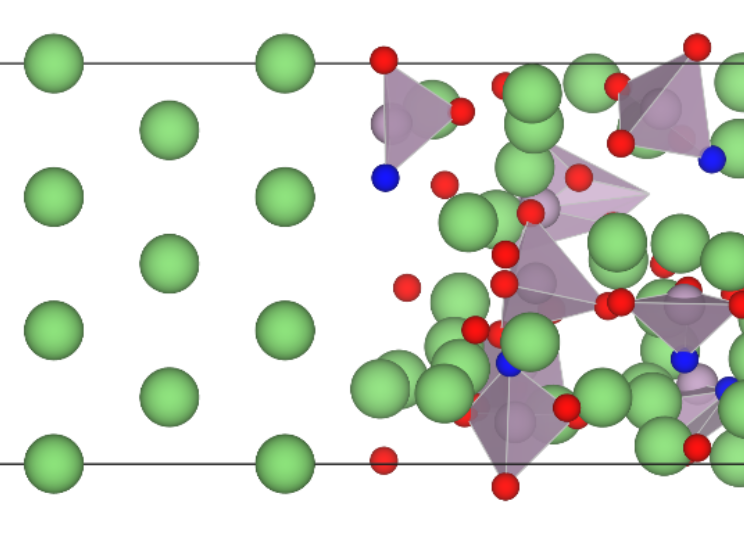
Investigating Li+ transport within the amorphous lithium phosphorous oxynitride (LiPON) framework, especially across a Li||LiPON interface, has proven challenging due to its amorphous nature and varying stoichiometry, necessitating large supercells and long timescales for computational models. Notably, machine learned interatomic potentials (MLIPs) can combine the computational speed of classical force fields with the accuracy of density functional theory (DFT), making them the ideal tool for modelling such amorphous materials. Thus, in this work, we train and validate the neural equivariant interatomic potential (NequIP) framework on a comprehensive DFT-based dataset consisting of 13,454 chemically relevant structures to describe LiPON. With an optimized training (validation) energy and force mean absolute errors of 5.5 (6.1) meV/atom and 13.2 (13.6) meV/Å, respectively, we employ the trained potential in model Li-transport in both bulk LiPON and across a Li||LiPON interface. Amorphous LiPON structures generated by the optimized potential do resemble those generated by ab initio molecular dynamics, with N being incorporated on non-bridging apical and bridging sites. Subsequent analysis of Li+ diffusivity in the bulk LiPON structures indicates broad agreement with computational and experimental literature so far. Further, we investigate the anisotropy in Li+ transport across the Li(110)||LiPON interface, where we observe Li-transport across the interface to be one order-of-magnitude slower than Li-motion within the bulk Li and LiPON phases. Nevertheless, we note that this anisotropy of Li-transport across the interface is minor and do not expect it to cause any significant impedance buildup. Finally, our work highlights the efficiency of MLIPs in enabling high-fideling modelling of complex non-crystalline systems over large length and time scales.