Abstract
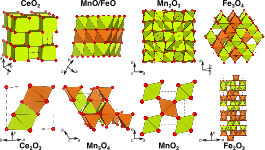
Using the strongly constrained and appropriately normed (SCAN) and SCAN+U approximations for describing electron exchange correlation (XC) within density functional theory, we investigate the oxidation energetics, lattice constants, and electronic structure of binary Ce, Mn, and Fe oxides, which are crucial ingredients for generating renewable fuels using two-step, oxide-based, solar thermochemical reactors. Unlike other common XC functionals, we find that SCAN does not overbind the O2 molecule, based on direct calculations of its bond energy and robust agreement between calculated formation enthalpies of main group oxides versus experiments. However, in the case of transition-metal oxides, SCAN systematically overestimates (i.e., yields too negative) oxidation enthalpies due to remaining self-interaction errors in the description of their ground-state electronic structure. Adding a Hubbard U term to the transition-metal centers, where the magnitude of U is determined from experimental oxidation enthalpies, significantly improves the qualitative agreement and marginally improves the quantitative agreement of SCAN+U-calculated electronic structure and lattice parameters, respectively, with experiments. Importantly, SCAN predicts the wrong ground-state structure for a few oxides, namely, Ce2O3, Mn2O3, and Fe3O4, while SCAN+U predicts the right polymorph for all systems considered in this paper. Hence, the SCAN+U framework, with an appropriately determined U, will be required to accurately describe ground-state properties and yield qualitatively consistent electronic properties for most transition-metal and rare-earth oxides.