Abstract
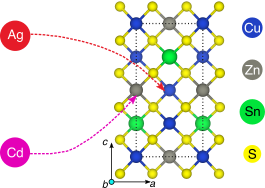
Cu2ZnSnS4-based solar cells, which constitute an inexpensive, beyond-Si photovoltaic technology, often suffer from low open-circuit voltage and efficiency. This drawback is often attributed to disorder in the Cu–Zn sublattice of the kesterite structure. While previous experiments have reported improved performances with isovalent substitution of Cd and Ag for Cu and Zn, respectively, the fundamental driving force for such improvements remains unclear. Here, we use density functional theory to study bulk stability, defect, and surface energetics, as well as the electronic structure of these dopants in Cu2ZnSnS4. We find that Cd and Ag can increase efficiencies, depending on the dopant concentration and Cu content used during synthesis. Most importantly, we find that a low level of Cd doping can suppress disorder in the kesterite phase across all Cu concentrations, while a low level of Ag doping can do so only when Zn- and Sn-rich conditions are employed. A higher Ag content is beneficial as it stabilizes the kesterite structure, whereas a higher Cd content is detrimental as it stabilizes the lower-gap stannite structure. Cd does not significantly influence the surface energetics of kesterite Cu2ZnSnS4. Ag, on the other hand, decreases the surface energies significantly, which would favor smaller particle sizes. We thus attribute the coarsening of particle size and changes in morphology observed during Cu2ZnSnS4 synthesis to annealing conditions during sulfurization and selenization, instead of any effect of Cd or Ag. Finally, we suggest the exploration of abundant, nontoxic, isovalent dopants, i.e., different from the attributes of Cd and Ag, to improve the performance of Cu2ZnSnS4.