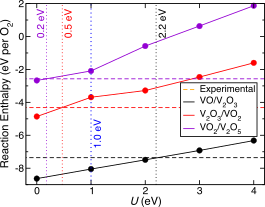
Abstract
Redox-active 3d transition-metal oxides (TMOs) are crucial ingredients for multiple sustainable energy applications, including solar cells, batteries, catalysis, and solar thermochemical water splitting. However, any predictive modeling, such as that employing density functional theory, needs to describe accurately the energetics of redox reactions involving transition metals, if new candidate materials are to be identified in a reliable fashion. Recently, we demonstrated that the state-of-the-art, strongly constrained and appropriately normed (SCAN) exchange-correlation functional requires a Hubbard U correction (determined, e.g., from experimental oxidation enthalpies) to reproduce the ground-state structure, lattice parameters, magnetic moments, and electronic properties of Ce-, Mn-, and Fe-based oxides. In the present work, we extend our approach to identify optimal U values for other 3d TMOs, specifically Ti, V, Cr, Co, Ni, and Cu, within the SCAN+U framework. We determine optimal U values of 2.5, 1.0, 3.0, and 2.5 eV for Ti, V, Co, and Ni oxides, respectively, while Cr and Cu oxides best reproduce redox thermodynamics without any U correction at all. While the U values required for Ti, V, Co, and Ni are lower than those needed within the generalized gradient approximation (GGA) + U or local density approximation (LDA) +U approaches, inclusion of U makes non-negligible improvements in ground-state property evaluations of these oxides. Here we also validate our optimal U values by performing a number of transferability checks for each 3d material. A SCAN+U framework (with an appropriately determined U) therefore is needed to assess accurately the ground-state energies and qualitatively consistent electronic structures for (most) first-row TMOs.