Abstract
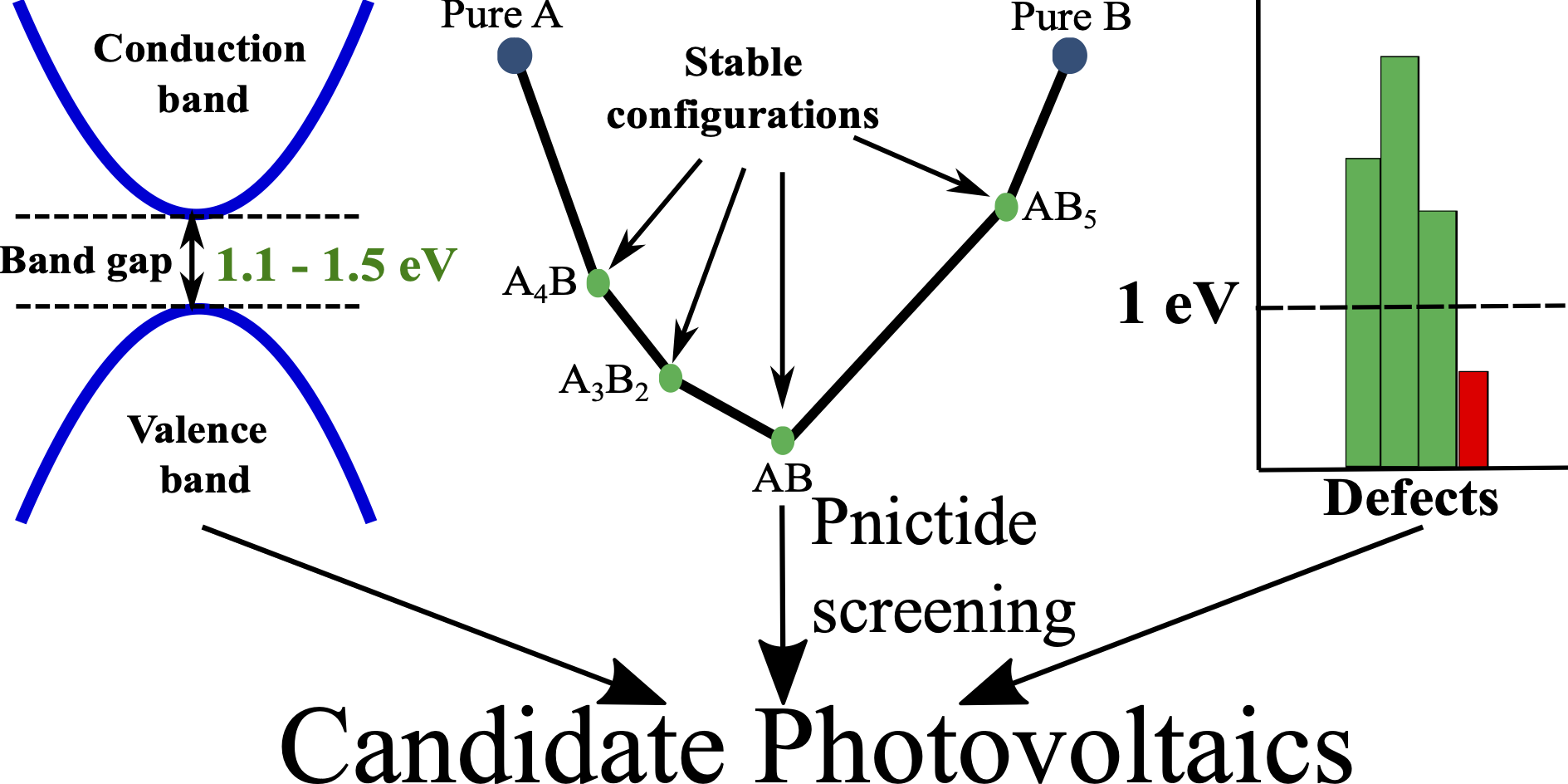
For the transition into a sustainable mode of energy usage, it is important to develop photovoltaic materials that exhibit better solar-to-electricity conversion efficiencies, a direct optimal band gap, and are made of non-toxic, earth abundant elements compared to the state-of-the-art silicon photovoltaics. Here, we explore the non-redox-active pnictide chemical space, including binary A3B2, ternary AA′2B2, and quaternary AA′A′′B2 compounds (A, A′, A′′ = Ca, Sr, or Zn; B = N or P), as candidate beyond-Si photovoltaics using density functional theory calculations. Specifically, we evaluate the ground state configurations, band gaps, and 0 K thermodynamic stability for all 20 pnictide compositions considered, besides computing the formation energy of cation vacancies, anion vacancies, and cation anti-sites in a subset of candidate compounds. Importantly, we identify SrZn2N2, SrZn2P2, and CaZn2P2 to be promising candidates, exhibiting optimal (1.1-1.5 eV) hybrid-functional-calculated band gaps, stability at 0 K, and high resistance to point defects (formation energies >1 eV), while other possible candidates include ZnCa2N2 and ZnSr2N2, which may be susceptible to N-vacancy formation. We hope that our study will contribute to the practical development of pnictide semiconductors as beyond-silicon light absorbers.