Abstract
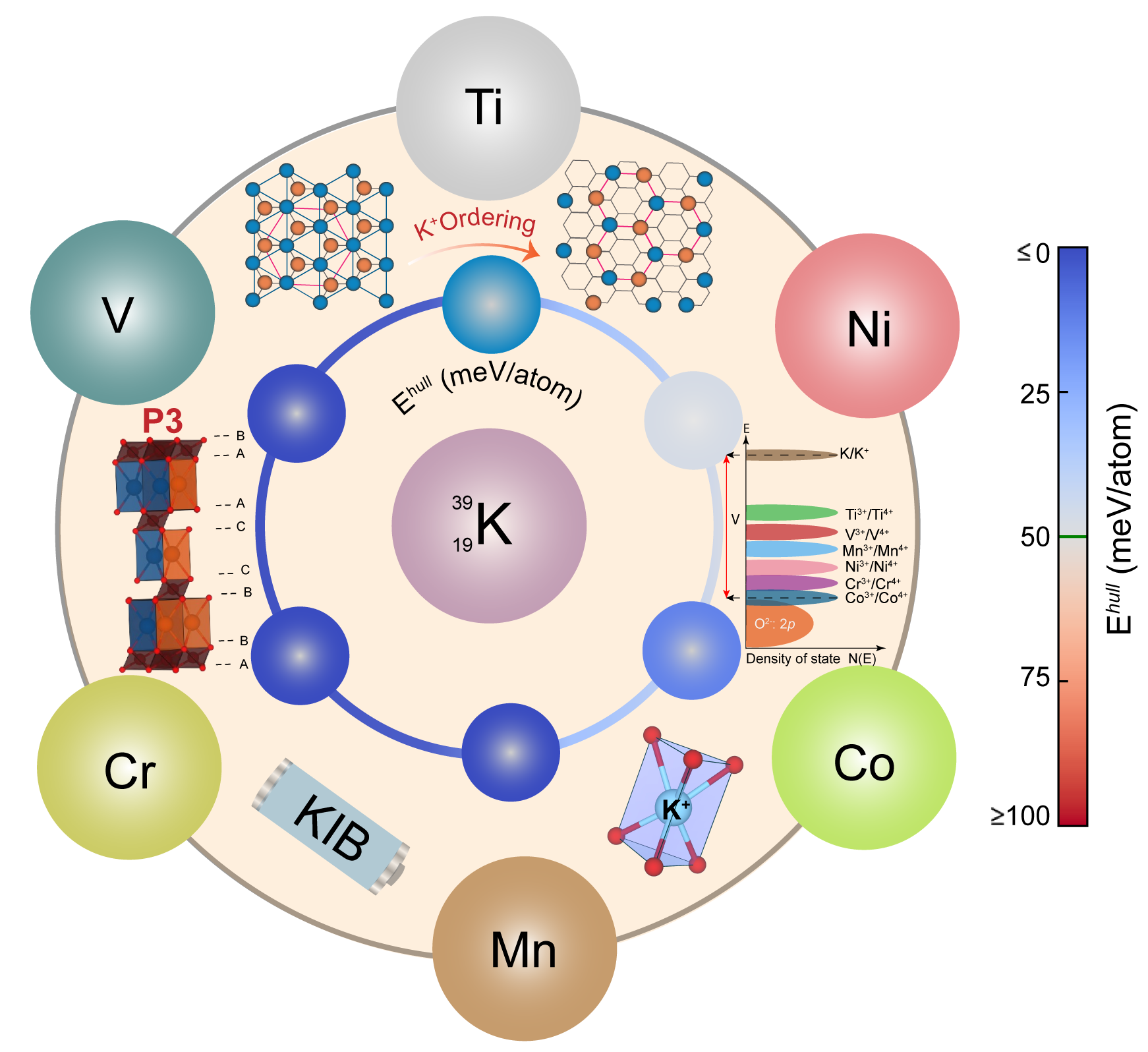
Given the increasing energy storage demands and limited natural resources of Li, K-ion batteries (KIBs) could be promising next-generation systems having natural abundance, similar chemistry, and energy density. Here, we have investigated the P3-type K0.5TMO2 (where TM = Ti, V, Cr, Mn, Co, or Ni) systems using density functional theory calculations as potential positive intercalation electrodes (or cathodes) for KIBs. Specifically, we have identified ground-state configurations and calculated the average topotactic voltages, electronic structures, on-site magnetic moments, and thermodynamic stabilities of all P3-K0.5TMO2 compositions and their corresponding depotassiated P3-TMO2 frameworks. Additionally, we evaluated the dynamic stability and K-mobility in select P3 structures. We find that K adopts the honeycomb or zig-zag configuration within each K-layer of all P3 structures considered, irrespective of the transition-metal (TM). In terms of voltages, we find the Co- and Ti-based compositions to exhibit the highest (4.59 V vs. K) and lowest (2.24 V) voltages, respectively, with the TM contributing to the redox behavior upon K (de-)intercalation. We observe all P3-K0.5TMO2 to be (meta)stable and hence experimentally synthesizable according to our 0 K convex hull calculations, while all depotassiated P3-TMO2 configurations are unstable and may appear during electrochemical cycling. Also, we verified the stability of the prismatic coordination environment of K compared to octahedral coordination at the K0.5TMO2 compositions using Rouxel and cationic potential models. Finally, combining our voltage and stability calculations, we find P3-KxCoO2 to be the most promising cathode composition, while P3-KxNiO2 is worth exploring. We also find P3-KxMnO2 to be worth pursuing given its dynamic stability and facile migration of K+ at both potassiated and depotassiated compositions. Our work should contribute to the exploration of strategies and materials required to make practical KIBs.