Abstract
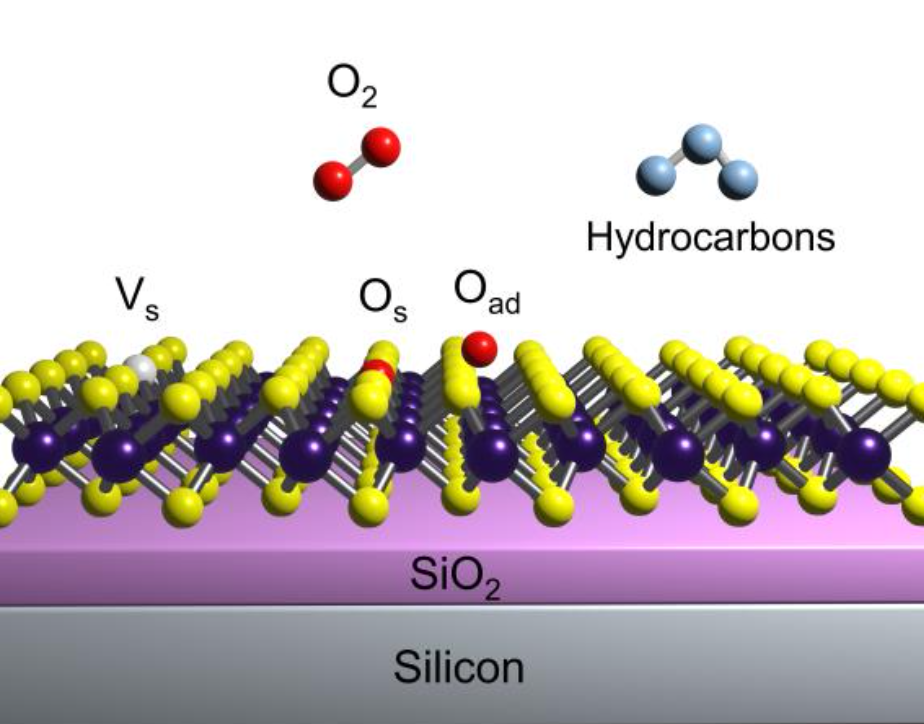
An intense low-energy broad luminescence peak (L-peak) is usually observed in 2D transition metal dichalcogenides (TMDs) at low temperatures. L-peak has earlier been attributed to bound excitons, but its origins are widely debated with direct consequences on optoelectronic properties. To decouple the contributions of physisorbed and chemisorbed oxygen, organic adsorbates, and strain on L-peak, we measured a series of monolayer (ML) MoS2 samples (mechanically exfoliated (ME), synthesized by oxygen-assisted chemical vapour deposition (O-CVD), hexagonal boron nitride (hBN) covered and hBN encapsulated). Emergence of L-peak below 150 K and saturation of photoluminescence (PL) intensity with laser power confirm bound nature of L-peak. Anomalously at room temperature, O-CVD samples show high A-exciton PL (c.f. ME), but reduced PL at low temperatures, which is attributed to strain-induced direct-to-indirect bandgap change in low defect O-CVD MoS2. Further, L-peak redshifts dramatically ~130 meV for O-CVD samples (c.f. ME). These observations are fully consistent with our predictions from density functional theory (DFT) calculations, considering effects of both strain and defects, and supported by Raman spectroscopy. In ME samples, charged oxygen adatoms are identified as thermodynamically favourable defects which can create in-gap states, and contribute to the L-peak. The useful effect of hBN is found to originate from reduction of charged oxygen adatoms and hydrocarbon complexes. This combined experimental-theoretical study allows an enriched understanding of L-peak and beneficial impact of hBN, and motivates collective studies of strain and defects with direct impact on optoelectronics and quantum technologies.